Compartilhar:



Síndrome de Pallister-Killiam Sintomas, causas, tratamento
O Síndrome de Pallister-Killian (SPK), também conhecida pelo nome de tetrasomia 12é uma doença rara, de origem genética, caracterizada por um amplo espectro de envolvimento de múltiplos órgãos.
Clinicamente, esta condição é definida por retardo mental, retardo psicomotor, hipotonia, fenótipo facial atípica, anomalias pigmentares da pele e alopecia (Toledo-Bravo Laguna, Campo-Casanelles, Santana-Rogriguez, Santana -Artiles, Sebastían-Garcñua, Cabrera-López, 2014).
Além disso, outros tipos de complicações médicas também podem aparecer relacionadas a malformações em diferentes sistemas corporais ou episódios convulsivos (Toledo-Bravo de la Laguna et al., 2014).
A origem etiológica dessa doença está associada a uma alteração genética distribuída em mosaico. Especificamente, é devido à presença de um cromossomo extra 12 em algumas células do organismo (Understanding Chromosome Disorders, 2016).
O diagnóstico da síndrome de Pallister-Killiam pode ser feito nos estágios pré e pós-natal. O objetivo principal é a identificação de características clínicas e o uso de um estudo genético de confirmação (Méndez, Rodríguez, Boluarte, Cartolin, Valdéz e Matheus, 2013).
Esta síndrome tem uma alta taxa de mortalidade (Ramírez Ferández, García Cavazos, Sánchez Martínez, 2007). No entanto, a abordagem médica farmacológica e o tratamento reabilitativo podem trazer importantes benefícios na qualidade de vida e no estado clínico dos afetados (Méndez et al., 2013).
Características da síndrome de Pallister-Killiam
A síndrome de Pallister-Killiam (SPK) é um tipo de doença genética em mosaico. Nesse caso, a alteração cromossômica afeta apenas algumas células do organismo.
Algumas instituições, como a Genectis Home Reference (2016) classificam essa patologia dentro dos chamados transtornos do desenvolvimento.
Em um nível geral, os distúrbios do desenvolvimento ou distúrbios do desenvolvimento em seu termo internacional geralmente se referem a um amplo conjunto de alterações e anormalidades físicas e cognitivas. Tudo isso resulta em um desvio ou atraso significativo do desenvolvimento em relação aos padrões normais ou esperados (Instituto Nacional de Distúrbios Neurológicos e Stroke, 2015).
Na síndrome de Pallister-Killiam, uma ampla afetação de diferentes sistemas e organismos do corpo é identificada (Genectis Home Reference, 2016)
Caracteriza-se principalmente por retardo mental, hipotonia muscular, desenvolvimento características faciais distintivas, pigmentação anormal da pele ou o crescimento do cabelo, entre outras anormalidades congênitas (Genectis Home Reference, 2016).
Além disso, a síndrome de Pallister-Kiliam é uma rara doença congênita origem (Turleau, 2009) que pode receber um grande número de nomes na literatura médica (Organização Nacional de Doenças Raras, 2016):
- Síndrome do mosaico de Pallister-Killiam.
- Síndrome do isquromossomo 12p.
- Síndrome de Killiam.
- Síndrome de Nicola-Teschler
- Síndrome do mosaico de Pallister.
- Tetrassomia 12p.
- Síndrome de Killiam-Tescheler-Nicola.
Esta doença foi inicialmente descrita por Pallister em 1977 (Toledo-Bravo de La Laguna et al., 2014).
Nas primeiras publicações, observou dois casos de pacientes adultos cujo curso foi caracterizado por várias conclusões: convulsões, hipotonia muscular, atraso mental, malformações músculo-esquelético e configuração facial orgânico, grosseiro e mudanças na cor da pele (Mendez et al., 2013).
Paralelamente, Teschler-Nicola e Killiam em 1981 descreveram esse mesmo quadro clínico em uma menina de três anos de idade (Méndez et al., 2013).
Por isso, nos primeiros relatórios clínicos, refira-se amplamente a uma condição médica caracterizada pela combinação de convulsões, atraso mental, e um fenótipo característica física (Toledo-Bravo Laguna et al., 2014).
Além disso, e em 1985 ele conseguiu identificar Gilgenkratz primeiro caso durante a gravidez, algo comum hoje graças a modernas técnicas de diagnóstico (de 2013 Mendez et al.).
Estatísticas
Os números de prevalência da síndrome de Pallister-Killiam não são conhecidos com precisão. Não foram feitos muitos diagnósticos definitivos e a maioria deles não foi publicada na literatura médica (Understanding Chromosome Disorders, 2016).
Assim, todos os autores e instituições definem esta síndrome como uma patologia genética rara ou rara na população geral (EuRed, 2016).
Cerca de 15 anos atrás, a síndrome de Pallister-Killiam havia sido identificada em apenas cerca de 100 casos em todo o mundo. Atualmente, esse número ultrapassou os 200 afetados (Understanding Chromosome Disorders, 2016).
Investigações epidemiológicas estimaram a incidência desta doença em cerca de 5,1 casos por milhão de crianças recém-nascidas (Understanding Chromosome Disorders, 2016), embora autores como Toledo-Bravo de la Laguna e colaboradores (2014) a classifiquem em 1 / 25.000
A maior prevalência associada às características sociodemográficas dos afetados não foi identificada. A síndrome de Pallister-Killian pode aparecer em qualquer gênero ou grupo técnico e / ou racial.
Signos e sintomas
No curso clínico da síndrome de Pallister-Killian, uma grande variedade de sinais e sintomas pode ser identificada. Todos eles associados a anomalias craniofaciais e / ou músculo esquelético e alterações cognitivas.
Configurações faciais
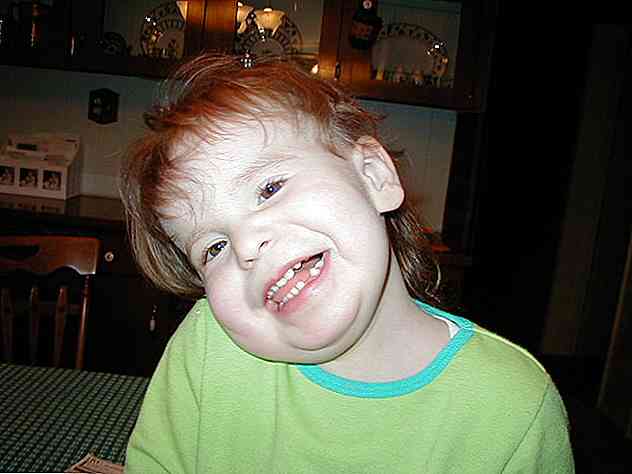
O desenvolvimento de malformações cranio-faciais, desde a fase de gestação até o crescimento pós-natal e infantil, constitui um dos sinais médicos mais característicos da síndrome de Pallister-Killiam.
Os sinais e sintomas mais comuns incluem anormalidades nas diferentes estruturas craniais e faciais que darão origem a uma aparência áspera e atípica (Toledo-Bravo de la Laguna et al., 2014; Entendendo Distúrbios Cromossômicos, 2016):
- Braquicefalia: Este termo refere-se a uma configuração craniana que resulta em aumento da largura da cabeça e achatamento das áreas occipital e posterior.
- Configuração craniana frontal: As áreas anterior e frontal da cabeça tendem a se desenvolver mais do que o normal. Uma testa proeminente ou protuberante pode ser vista.
- Configuração craniana posterior: a área mais posterior da cabeça soa para apresentar um estado subdesenvolvido. Um occipital plano pode ser visto.
- Hiperterismo: os olhos devem ser colocados a uma distância maior que o normal. Em um nível visual, os olhos são amplamente separados.
- Configuração nasal: O nariz geralmente tem um grande volume, com uma raiz ou ponte larga. As narinas devem ser orientadas para a frente (narinas antevertidas).
- Configuração oral e maxilar: as estruturas orais têm que apresentar um tamanho anormal. A mandíbula é menor que o habitual (micrognatia). O lábio superior adquire uma aparência fina e reduzida, enquanto o lábio inferior é espesso. A língua é maior que o esperado e o sulco nasolabial longo.
- Pavilhões auditivos: as orelhas têm uma posição baixa e são giradas para trás.
- Alopecia:O crescimento do cabelo é anormal em diferentes áreas. O mais comum é observar pequenas áreas de calvície nas sobrancelhas, cílios ou cabeça.
- Pontos acrômicos e hipercrômicos: É possível identificar o desenvolvimento de pequenas manchas nas áreas faciais. Eles são caracterizados pela perda de cor ou aparência escura.
Malformações musculoesqueléticas
Apesar de ser menos significante que as alterações faciais, é muito comum observar diversas anormalidades musculoesqueléticas em pacientes com Síndrome de Pallister (Understanding Chromosome Disorders, 2016):
- Pescoço: A distância entre a cabeça e o tronco do corpo é geralmente reduzida. Em um nível visual, podemos ver um pescoço curto ou menor que o normal.
- Coluna vertebral: Embora não seja muito comum identificar alterações na coluna vertebral, é possível que espinha bífida, apêndice sacro, escoliose ou cifose possam aparecer.
- Dicas: Os braços e pernas também mostram um crescimento anormal, sendo menor que o esperado para o sexo e a idade biológica da pessoa afetada.
- Polidactilia: alterações relacionadas ao número de dedos das mãos e dos pés também podem aparecer. O mais comum é ver mais dedos nas mãos
Hipotonia muscular e retardo psicomotor
Anormalidades relacionadas à estrutura e mobilidade muscular são outra das principais características clínicas da síndrome de Pallister-Killian (Understanding Chromosome Disorders, 2016):
A hipotonia muscular refere-se à identificação de um tônus ou tensão muscular anormalmente reduzida. Em um nível visual, a flacidez e a labilidade podem ser observadas em diferentes grupos musculares, especialmente acentuados nas extremidades.
Assim, a patologia muscular e esquelética causará um atraso significativo na aquisição de diferentes habilidades motoras, tanto no período neonatal como no período da infância.
Embora os períodos de desenvolvimento sejam variáveis entre os afetados, o calendário mais comum inclui os seguintes marcos:
- Sedestación: a capacidade de adquirir posturas independentemente, sentar ou girar com seu próprio corpo pode começar a desenvolver a partir de 3 meses. No entanto, em pessoas afetadas por esta síndrome pode ser adiada até os 8 anos de idade.
- Primeiros passosA coisa normal é que as crianças começam a dar seus primeiros passos em torno de 12 meses, no entanto, nesta patologia, este marco evolutivo pode ser adiado até os 9 anos. Além disso, em muitos casos, alguns métodos compensatórios, como talas ou calçados especializados, são essenciais.
Alterações neurológicas
Outra das áreas fortemente afetadas é o sistema nervoso. Na maioria dos casos, os sinais e sintomas estão relacionados principalmente a convulsões e deficiência intelectual (Toledo-Bravo de la Laguna et al., 2014; Entendendo Distúrbios Cromossômicos, 2016):
- Convulsões: a presença e o desenvolvimento de uma atividade elétrica neuronal incomum, alterada e desorganizada pode levar à presença de eventos recorrentes definidos por espasmos musculares, agitação motora ou ausência de consciência. A estrutura cerebral está gravemente comprometida, levando a uma significativa deterioração cognitiva e tecidual.
- Discapacidade intelectual: Embora o nível de comprometimento cognitivo seja variável, na maioria dos casos é identificado um quociente intelectual baixo ou limítrofe. As áreas mais afetadas são as psicomotoras e linguísticas, com os critérios clínicos de transtorno do espectro autista preenchendo uma das áreas afetadas.
- Atraso generalizado no desenvolvimento: o ritmo de aprendizado das diferentes habilidades diárias e acadêmicas é geralmente lento em boa parte dos afetados. Adaptações e apoio escolar especializado são normalmente necessários.
Outras anomalias
Embora sejam menos frequentes, outros tipos de complicações médicas também podem aparecer (Organização Nacional para Desordens Raras, 2016) (Toledo-Bravo de la Laguna et al., 2014):
- Anormalidades e malformações cardíacas, gastrointestinais, renais e genitais.
- Estenose auditiva.
- Hipoplasia pulmonar.
- Estrabismo e catarata.
- Redução da acuidade visual e auditiva.
Causas
A origem da síndrome de Pallister-Killian está associada a uma anormalidade genética em mosaico no cromossomo 12. Ela afeta apenas o material genético de algumas células do organismo (Inage et al., 2010).
Os cromossomos são parte do núcleo de todas as células encontradas no corpo humano. Eles são compostos de uma ampla variedade de componentes bioquímicos e contêm a informação genética de cada indivíduo (National Organization for Rare Disorders, 2016).
Os humanos têm 46 cromossomos diferentes, organizados em pares e numerados de 1 a 23. Além disso, no nível individual, cada cromossomo tem uma área ou braço curto chamado "p" e outro longo chamado "q" (Organização Nacional para Desordens Raras, 2016).
A anomalia afeta o cromossomo 12 e leva à presença de um cromossomo com estrutura anormal, chamado isocromossomo (Genetics Home Reference, 2016).
Assim, esse cromossomo tende a ter dois braços curtos em vez de um de cada configuração p (curto) e longo (q) (Genetics Home Reference, 2016).
Como conseqüência, a presença de material genético extra e / ou anômalo irá alterar o curso normal e eficiente do desenvolvimento físico e cognitivo da pessoa afetada, dando origem às características clínicas da síndrome de Pallister-Killian (Genetics Home Reference, 2016). .
Diagnóstico
A síndrome de Pallister-Killian pode ser identificada durante a gravidez ou no estágio pós-natal, com base nas características clínicas e nos resultados de diferentes testes laboratoriais (Turleau, 2009).
Durante a gestação, os exames mais utilizados são ultrassonografia ultrassônica, amniocentese ou biópsia de vilosidades coriônicas (Turleau, 2009).
Nesse sentido, a análise do material genético do embrião pode nos oferecer uma confirmação dessa patologia, através da identificação de anomalias compatíveis (Turleau, 2009).
Por outro lado, se o diagnóstico é feito após o nascimento, é fundamental (Understanding Chromosome Disorders, 2016):
- Biópsia Cutânea.
- Análise de sangue
- Estudo de linfócitos sanguíneos.
- Hibridização fluorescente in situ.
- Hibridização genômica comparativa.
Tratamento
Nenhuma terapia específica foi desenvolvida para o tratamento de pessoas com síndrome de Pallister-Killian (National Organization for Rare Disorders, 2016).
A síndrome de Pallister-Killian geralmente está associada a um mau prognóstico e altas taxas de mortalidade (Ramírez Ferández, García Cavazos, Sánchez Martínez, 2007).
No entanto, o tratamento reabilitador, a educação especial e a terapia ocupacional podem oferecer um bom prognóstico funcional e um aumento na qualidade de vida das pessoas afetadas.
Por exemplo, Méndez e sua equipe (2013) descrevem um caso de tratamento reabilitativo caracterizado por:
- Melhorias nas habilidades psicomotoras: controle da cabeça, sentado e em pé independente.
- Melhoria no nível de atenção, atenção, regulação comportamental.
- Melhoria das habilidades motoras finas, como a pressão manual.
- Emissão de sons e sorriso contextual.
- Rastreamento visual, fixação e discriminação de estímulos auditivos.
Referências
- Ecured (2016). Síndrome de Pallister-Killian. Obtido de Ecured.
- Genetics Home Reference. (2016). Síndrome do mosaico de Pallister-Killian. Obtido a partir da Genetics Home Reference.
- Inage et al. (2010). Sobreposição fenotípica da trissomia 12p e síndrome de PallistereKillian. Revista Européia de Genética Médica, 159-161.
- Méndez, M., Rodríguez, M., Boularte, A., Cartolin, R., Valdéz, G., & Matheus, F.(2013). Síndrome de Killian-Pallister. Relato de um caso em terapia de reabilitação interdisciplinar. Rev Med Hered.
- NORD (2016.) Síndrome do mosaico de Pallister Killian. Retirado da Organização Nacional para Desordens Raras.
- Ramírez-Fernández, M., Garcia Cavazos, R., & Sánchez-Martínez, H. (2007). Síndrome de Pallister-Killiam. Comunicação de um caso. Ginecol e obsta Mex, 414-18.
- Toledo-Bravo de la Laguna, L., Campo-Casanelles, M., Santana-Rodriguez, A., Santana-Artiles, A., Sebastian-Garcia, I., e Cabrera-Lopez, J. (2014). Apresentação de três casos de síndrome de Pallister-Killiam. Rev Neurol, 63-68.
- Turleau, C. (2009). Tetrassomia 12p. Obtido da Orphanet.
- Entendendo Distúrbios Cromossômicos. (2016). Síndrome de Pallister-Killiam. Entendendo Distúrbios Cromossômicos.
- Imagem de origem.