Compartilhar:



Sintomas, causas e tratamentos da síndrome de Rokitnasky-Küster-Hauser
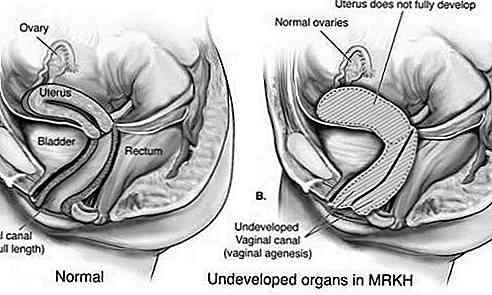
Síndrome de Rokitansky-Küster-Hauser (MRKH) é um distúrbio que afeta o sistema reprodutivo feminino caracterizado pelo subdesenvolvimento ou ausência do útero e da vagina.
As mulheres com esta síndrome desenvolvem características sexuais secundárias durante a puberdade - senes, pêlos púbicos - mas não apresentam um ciclo menstrual (amenorreia primária).
Não iniciar o ciclo menstrual é frequentemente o sinal inicial da síndrome de MRKH. Embora as mulheres com essa condição não possam engravidar, elas podem ter filhos por meio da reprodução assistida.
A gravidade da síndrome pode variar dependendo do tipo. O tipo 1 é caracterizado por uma ausência isolada dos dois terços proximais da vagina. Tipo II é caracterizada por defeitos outros, tais como anomalias renais (40% dos casos), anormalidades esqueléticas (20-25%), deficiência (10%) de audição, e mais raramente, defeitos cardíacos.
Devido à natureza da doença, é provável que cause problemas psicológicos significativos, por isso é recomendável pedir ajuda.
A síndrome de MRKH tem uma incidência mundial de 1 em cada 4500 nascimentos de mulheres, de acordo com vários estudos.
Geralmente é diagnosticado com maior frequência na adolescência, quando é verificado se o ciclo menstrual se desenvolve. Embora as mulheres não possam ter filhos, seus ovários são normais e funcionais.
Sintomas da síndrome de Rokitnasky-Küster-Hauser
Os sintomas da síndrome de MRKH variam muito de uma mulher para outra, portanto, tenha em mente que as mulheres afetadas podem não ter todos os sintomas mencionados abaixo.
Síndrome de Mayer-Rokitansky-Hauser tipo I
Este tipo também é conhecido como aplasia de Müller e é caracterizado pelo desenvolvimento inadequado do útero e da vagina. Na maioria dos casos, o útero e / ou a vagina não se desenvolveram (aplasia); ou há um estreitamento da parte superior da vagina e do útero (atresia). As trompas de Falópio também podem ser afetadas.
Algumas das características ou sintomas da síndrome de MRKH são:
-Primeira menopausa ou ausência de períodos durante a puberdade. Embora esse sintoma seja comum na síndrome de MRKH, o paciente é submetido à puberdade com telarca e adrenarca normais. No entanto, não há menstruação.
Como a função dos ovários é normal, os pacientes experimentam todas as mudanças associadas à menstruação ou à puberdade.
Órgãos genitais externos normais.
- Profundidade vaginal reduzida, de 2 a 7 cm.
- Características sexuais, como mamas normais e pêlos pubianos.
-Ovarios funcionais com níveis normais de estrogênio.
- Modelos cromossômicos normais.
-Na síndrome de MRKH tipo 1, apenas a vagina e o útero são anormais. No tipo II MRKH, defeitos na vagina e do útero pode também ser acompanhada por anormalidades na trompa de Falópio, como no rins ou na coluna vertebral.
-Embora as anormalidades cardíacas sejam raras, a janela aorta-pulmonar, a comunicação interatrial e a estenose valvar pulmonar podem ocorrer
-Pacientes com síndrome de MRKH geralmente se queixam de dor abdominal cíclica devido ao descolamento do endométrio cíclico sem uma via de drenagem patente.
-Esterilidade: Muitos pacientes freqüentemente procuram atendimento clínico para infertilidade, mas não para amenorréia primária.
- Outro sintoma é a dificuldade nas relações sexuais, uma vez que o grau de aplasia vaginal pode variar desde a ausência completa até o desenvolvimento deficiente, o que pode causar dispareunia.
-Dificuldades de micção, incontinência urinária, infecções urinárias recorrentes.
- anormalidades vertebrais.
Um sling de tecido palpável pode estar presente no nível da reflexão peritoneal.
- malformações renais.
Sintomas da síndrome MRKH tipo II
A insuficiência renal é a anomalia mais comum associada à síndrome de MRKH tipo II.
Mulheres com MRKH tipo síndrome II pode não ter nenhum rim, malformação de um ou ambos os rins (displasia renal), o desenvolvimento de rins (hipoplasia) e / ou colocação incorrecta dentro do corpo de um ou ambos os rins (ectopia renal).
Estas anomalias podem causar deficiência renal crescimento, pedras nos rins, aumento da susceptibilidade a infecções do tracto urinário e acumulação anormal de urina no rim devido a uma obstrução.
Muitas mulheres com síndrome MRKH tipo II pode também têm malformações esqueléticas, tais como problemas ósseos nas vértebras cervicais e torácicas que pode se desenvolver de forma inadequada.
Anormalidades da face também podem ocorrer, tais como ter uma anormalmente pequena mandíbula (micrognatia), lábio leporino, e subdesenvolvimento de um lado da face, resultando em assimetria facial.
Muitas mulheres afetadas também podem desenvolver problemas de audição, principalmente devido a anormalidades estruturais da orelha média.
Quando as orelhas estão envolvidas, o distúrbio pode ser chamado de Síndrome do Ouvido Renal Genital (GRES).
Algumas das mulheres com síndrome MRKH tipo II tiveram anormalidades físicas adicionais que incluem defeitos nas mãos e / ou braços.
Estas anormalidades nos membros podem incluir ausência de um ou mais dedos das mãos e pés, polegar duplicado e ausência de delgado longo antebraço (ausente raio) osso. Nem todos os sintomas ocorrem em todos os pacientes, depende da localização e gravidade.
Causas
Na maioria dos casos, a origem da síndrome de MRKH é desconhecida e ocorre em mulheres sem histórico familiar.
Os pesquisadores apontam para fatores genéticos e ambientais como as causas da síndrome, embora nenhum gene ou genes associados à doença tenham sido ainda determinados.
Historicamente, os pesquisadores têm sugerido que a síndrome pode ocorrer como resultado de doença ou exposição fetal materna a várias substâncias prejudiciais, tais como certas drogas, abuso de drogas, álcool ...
No entanto, não houve nenhum estudo que corrobore essa associação entre a síndrome e o consumo de algum outro medicamento.
Em algumas famílias, a síndrome parece ter um padrão de herança dominante. No entanto, o modelo de herança é difícil de estabelecer, uma vez que nem todas as mulheres que sofrem do mesmo apresentam os mesmos sintomas, mesmo que sejam da mesma família.
Segundo os pesquisadores, é mais provável que seja uma combinação de fatores genéticos e ambientais.
Quanto às anomalias no processo de reprodução, elas se devem a um desenvolvimento incompleto do ducto de Müller, mas sua causa permanece desconhecida.
Parece que nos últimos anos, houve um aumento na evidência, em que a síndrome de MKRH é uma desordem genética. O aumento nos estudos de caso significou que essa ideia continua a ser reforçada.
Estamos falando de desordens genéticas quando há uma combinação de genes para um traço particular. Se é um distúrbio genético dominante como grave neste caso, seria produzido pelo desenvolvimento anormal de uma única cópia de um gene anormal.
Esse gene defeituoso pode ser herdado tanto pela mãe quanto pelo pai ou pode ser devido a uma nova mutação no próprio gene.
A probabilidade, portanto, de transmitir esse gene defeituoso será de 50% para cada gravidez, independentemente do sexo da criança.
A herança poligênica também foi proposta como causa da síndrome de MRKH.
Até o momento, sete deleções e uma duplicação de segmentos cromossômicos foram encontradas em várias pessoas afetadas pela síndrome de MRKH. Mas apenas uma dessas anomalias foi encontrada por pessoa.
Atualmente, esses cromossomos foram identificados onde é possível que eliminações segmentais estejam envolvidas. Estes cromossomas são: cromossoma 1 (1q21.1), 4 (4q34q), 8 (8p23, 1), 10 (10p14-15), 16 (16p11.2), 17 (17q12) e 22 (22q11.21) e a duplicação foi encontrada no cromossomo X (Xpter-p22.32).
Toda esta nova informação levou os pesquisadores a selecionar vários genes candidatos, incluindo: HNF1B, Lhx1, TBX6, ITIH5 e SHOX.
Tratamento
O objetivo do tratamento é que o paciente tenha um funcionamento sexual completo e satisfatório.
Como os sintomas são tão variáveis, é necessária a colaboração de uma equipe de especialistas, a fim de garantir uma abordagem abrangente ao tratamento.
Como regra geral, os pacientes com síndrome de MRKH são encorajados a procurar ajuda psicológica após o diagnóstico e antes do tratamento, pois esta síndrome pode causar ansiedade ou sofrimento psicológico. Também são recomendados grupos de suporte.
Em relação ao tratamento da aplasia vaginal é criar uma nova vagina.
O tratamento pode ser cirúrgico ou não. O não-cirúrgico será sempre a primeira opção. Uma das opções são os dilatadores vaginais, que servem para ajudar a aumentar ou criar uma vagina.
O método mais conhecido é o dilatador de Franck, também conhecido como dilatador perineal, que não requer nenhum tipo de operação cirúrgica.
Sendo auto-administrado o paciente deve estar suficientemente motivado para usá-lo. Dura aproximadamente seis semanas a vários meses.
Outra opção pode ser uma vaginoplastia, que criaria uma vagina superficial. Mas sobre esta cirurgia não há muito acordo sobre quais técnicas usar.
A técnica de Mclndoe é um dos procedimentos cirúrgicos mais utilizados para a reconstrução da vagina. Um enxerto de pele retirado das nádegas ou da coxa é retirado e aplicado a uma prótese em forma de pênis inflável. Esta prótese enxerto com os moldes para túnel vaginal, que se abre com um bisturi e dissecados no corte de forma e sem corte, com cuidado para não danificar o vegija, recto ou peritoneu.
É deixado de sete a dez dias e, quando é trocado, está sob anestesia. Mais tarde, o paciente usará outras próteses de dilatação. Aos três meses, o paciente pode fazer sexo.
O problema com esta técnica é que, além de deixar cicatrizes, é necessária dilatação a longo prazo.
Outra técnica utilizada para a síndrome de MRKH é a neo-vagina intestinal.
Esta técnica usa um segmento isolado dos intestinos. Consiste em seccionar um fragmento do cólon sigmóide através de uma incisão abdominal e transferi-lo para a área onde o espaço da neovagina foi criado. Esta técnica é bastante complexa e pode durar até 8 horas.
A vantagem dessa técnica é que ela deixa uma vagina espaçosa de comprimento adequado e também é autolubrificante. Em relação às desvantagens, a primeira coisa é que é uma cirurgia complexa e infrabominal, deixa o excesso de secreções mucosas através do fragmento intestinal, prolapso vaginal mucoso e inflamação da mucosa na neovagina.
Finalmente, outra das técnicas é a Técnica Vecchieti.
Esta técnica exerce uma pressão progressiva contínua por uma azeitona acrílica através do poder do espaço neovaginal e da parede abdominal. Em seguida, um dispositivo de tração é colocado na cavidade peritoneal e, gradualmente, remove a abóbada vaginal. Esta técnica é agora feita através de laparoscopia.
Referências
- https://visualsonline.cancer.gov/
- https://ghr.nlm.nih.gov/condition/mayer-rokitansky-kuster-hauser-syndrome
- http://emedicine.medscape.com/article/953492-overview
- http://www.news-medical.net/health/Mayer-Rokitansky-Kuster-Hauser-(MRKH)-Syndrome-Symptoms-(Espanhol)).aspx
- http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=3109
- https://rarediseases.org/rare-diseases/mayer-rokitansky-kuster-hauser-syndrome/
- http://www.biomedcentral.com/content/pdf/1750-1172-2-13.pdf
- Imagem da fonte: http://www.mrkhnorge.org/mrkh/?lang=en