Compartilhar:



Síndrome de Tolosa-Hunt Sintomas, Causas, Tratamento
O Síndrome de Tolosa-Hunt é um tipo raro de oftalmoplegia dolorosa (Buedo Rubio, Martin-Tamayo Onsurbe Blazquez e Ramirez, 2015).
Clinicamente, a doença é caracterizada pela presença de episódios de dor ou preorbitario hemicraniano, paralisia oculomotor, anormalidades pupilares e hipoestesia ou hiperalgesia em várias áreas áreas (Martinez, Casasco, Pendre, De Bonis e Berner, 2010).
A origem do quadro clínico desta doença é devido ao desenvolvimento de lesões, em vários nervos cranianos: III, IV, IV e / ou V. Consequentemente causa inchaço do seio cavernoso, o ápice orbital e a fissura orbital superior (Martinez, Casasco, Pendre, De Bonis e Berner, 2010).
Entretanto, a causa etiológica desse processo patológico ainda não é conhecida exatamente. Geralmente não está relacionado a fatores primários ou secundários, como trauma, tumores, infecções, etc. (Granados Reyes, Soriano Redondo e Durán Ferreras, 2012).
Além do exame físico no diagnóstico de síndrome de Tolosa-Hunt geralmente seguido os critérios propostos pela Classificação Internacional Dores de cabeça Doenças Segunda Edição (Diaz, aedo e González Hernández, 2009).
Neste caso, o teste de escolha para identificar distúrbios do nervo é a ressonância magnética nuclear (RMN) (Diaz, Aedo e González Hernández, 2009).
O tratamento padrão da síndrome de Tolosa Hunt baseia-se na administração de medicação corticóide oral (Zimmermann Paiz, 2008).
Características da síndrome de Tolosa-Hunt
A síndrome de Tolosa-Hunt é uma oftalmoplegia caráter dolorosa causada pela inflamação idiopática de diversas áreas oculares e orbitais (Paiz Zimmermann, 2008).
O termo oftalmoplegia dolorosa utilizado no campo médico e experimental para se referir a uma condição definida por (Martinez, Casasco, Pendre, De Bonis e Berner, 2010):
- Dor localizada em regiões orbitais ou cranianas (especialmente no nível unilateral).
- Paralisia Ipsilateral dos músculos oculomotores.
- Anormalidades na contração pupilar.
- Alterações de sensibilidade em várias áreas faciais.
Este quadro clínico é geralmente causada por uma ampla variedade de processos entre os quais acontecimentos traumáticos, doença vascular, tumores, processos infecciosos ou inflamatórios, neuropatia diabética, a enxaqueca, etc. (Serralta San Martín, Torrecillas Narvaez, Soler Rangel, Ibáñez Sanz e Gómez Cerezo, 2013).
No caso de síndrome de Tolosa Hunt, embora o processo patológico, que dá origem ao seu curso clínico não é exatamente conhecida, as áreas mais afetadas são Martínez, Casasco, Pendre, De Bonis e Berner, 2010):
- Seio cavernoso: os seios cavernosos localizam-se no nível endocraniano, circundados pela dura-máter entre o apêndice orbitariano e a fissura orbital superior. É um plexo venoso, isto é, um grupo venoso de paredes finas que tem que formar uma cavidade. No seu interior contém a artéria carótida interna e algumas partes cranianas (III, IV, V, etc.).
- Apx orbital: refere-se ao ápice do canal orbital através do qual o nervo óptico passa, fibras do plexo coróide e artéria oftálmica.
- Fissura Orbital Superior: esta estrutura está localizado na ranhura formando a maior e menor área do osso esfenóide (localizado na área interna da face em limites subsequentes de órbitas oculares. Ele contém o ramo inferior e superior do crânio II, IV e VI (inervar os músculos oculares extras).
Essa síndrome foi inicialmente descrita pelo neurocirurgião espanhol Eduardo Tolosa em 1954 (Granados Reyes, Soriano Redondo e Durán Ferreras, 2012).
Na descrição inicial, Tolosa se referiu a um paciente cujo quadro clínico é caracterizado por:
- Dor orbital na área esquerda.
- Oftalmoplegia ipsilateral.
- Perda significativa de acuidade visual.
- Hipossesia no primeiro ramo do nervo trigêmeo.
A autópsia revelou uma inflamação granulomatosa do seio cavernoso (Martinez, Casasco, Pendre, De Bonis e Berner, 2010).
Anos depois, em 1982, William Hunt definiu um total de 6 casos semelhantes. Além disso, ele conseguiu demonstrar a eficácia do tratamento com corticosteróides na melhoria dos sintomas associados (Martinez, Casasco, Pendre, De Bonis e Berner, 2010).
Smith e Taxal foram os primeiros autores a nomear essa entidade clínica como síndrome de Tolosa Hunt em 1966 (Martínez, Casasco, Pendre, De Bonis e Berner, 2010).
Atualmente, os critérios clínicos e diagnósticos para essa síndrome foram categorizados pela International Headache Society em 2004 (Martínez, Casasco, Pendre, De Bonis e Berner, 2010).
É uma patologia frequente?
A síndrome de Tolosa Hunt é uma doença rara na população geral (National Organization for Rare Disorders, 2016).
Alguns autores, como Taylor (2015), apontam que é incomum nos Estados Unidos e internacionalmente, embora dados precisos sobre sua prevalência não sejam conhecidos.
Registros clínicos mostram que a síndrome de Tolosa Hunt é responsável por cerca de 9% das condições inflamatórias orbitárias (Zimmermann Paiz, 2008).
Assim, estima-se que sua incidência seja de 1-2 casos por milhão de pessoas no mundo (Aguirre, Zúñiga e Barrera, 2014).
É apresentado de forma equivalente em homens e mulheres, com uma idade média de início de 41 anos (National Organization for Rare Disorders, 2016).
No entanto, alguns casos de apresentação antecipada em pessoas com menos de 30 anos foram documentados. Em alguns casos esporádicos, esta síndrome pode afetar crianças menores de 10 anos de idade (National Organization for Rare Disorders, 2016).
Signos e sintomas
síndrome de STH, caracterizada principalmente pela presença de um oftalmologia doloroso (Martinez, Casasco, Pendre, De Bonis e Berner, 2010).
Os sinais e sintomas mais comuns estão relacionados à presença de episódios de dor, inflamação, parestesias, paralisia, etc.
Inflamação orbital
Como referido acima, um dos sinais característicos do síndrome de TolosaHunt é a inflamação do seio cavernoso, ápice orbital e a fissura orbital superior (Martinez, Casasco, Pendre, De Bonis e Berner, 2010).
No nível visual, é identificada uma inflamação significativa de todas as áreas faciais ao redor dos globos oculares (National Organization for Rare Disorders, 2016).
Também é possível identificar (National Organization for Rare Disorders, 2016):
- Proptose: profusão de globos oculares secundários à inflamação anatômica de estruturas adjacentes.
- Ptose: flacidez e queda das pálpebras superiores secundárias a uma afetação nervosa.
Dor orbital
As pessoas afectadas muitas vezes sofrem episódios recorrentes de dor aguda localizada em áreas orbitais e faciais perto dos olhos (Martinez, Casasco, Pendre, De Bonis e Berner, 2010).
Isso geralmente é unilateral, afeta um lado do rosto, embora tenha havido casos com envolvimento bilateral (Granados Reyes, Soriano Redondo e Durán Ferreras, 2012).
É freqüente que a dor se expanda progressivamente para as áreas frontal e temporal. Pode durar até 8 semanas na ausência de uma abordagem terapêutica eficiente (Martínez, Casasco, Pendre, De Bonis e Berner, 2010).
Além disso, episódios de dor são descritos por pessoas afetadas como (Granados Reyes, Soriano Redondo e Durán Ferreras, 2012):
- Intenso
- Urentes.
- Lacerantes
- Socos
Pode ser acompanhada de outros sintomas, tais como oculomotor parestesia dentro das primeiras poucas semanas após o início (Martinez, Casasco, Pendre, De Bonis e Berner, 2010).
Este tipo de dor deriva fundamentalmente de uma neuralgia do ramo oftálmico do nervo craniano V e do ramo maxilar. No entanto, o emprego pode reduzir ou eliminar a dor nas primeiras 24 horas (Martínez, Casasco, Pendre, De Bonis e Berner, 2010).
Paresia oculomotora
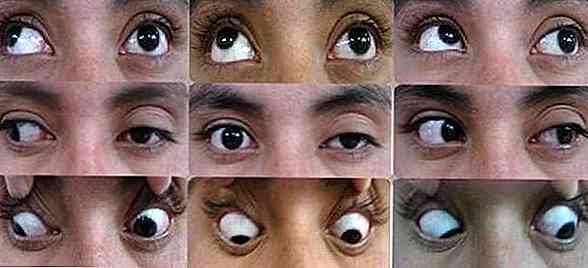
Um dos achados centrais da síndrome de Tolosa Hunt é a presença de paresia, ou seja, ausência ou diminuição significativa do movimento voluntário.
Neste tipo de patologia, a fraqueza muscular e / ou paralisia parcial afetam preferencialmente as áreas oculomotoras.
O mais comum é que episódios de paresia que afetam os nervos cranianos são identificados (Sánchez Iñigo e Navarro González, 2014).
- III (nervo oculomotor): controla tanto a contração pupilar quanto o movimento dos globos oculares.
- IV (nervo troclear): é responsável pelo controle da função motora do músculo ocular oblíquo superior
- VI (nervo Abducens): é responsável por controlar as funções motoras oculares externas, ou seja, o movimento gerado pelo músculo reto lateral.
Em um nível visual, podemos observar como os afetados são incapazes de realizar atos motores com os olhos. Eles não são capazes de mover os olhos ou direcionar o olhar para direções diferentes (National Organization for Rare Disorders, 2016).
Distúrbios pupilares
Apesar de não estar presente em todos os casos, em alguns afetados podemos identificar alterações relacionadas à função pupilar.
Nestes casos, uma das disfunções mais comuns é a midríase ou a miose pupilar.
- Midríase: aumento anormal do tamanho e diâmetro da pupila. Dilatação exagerada ocorre.
- Miose: diminuição anormal do tamanho e diâmetro da pupila. Há uma contração exagerada.
Anomalias visuais
As anormalidades oculares e orbitais descritas acima podem resultar em uma redução variável na acuidade visual (National Organization for Rare Disorders, 2016).
Causas
sinais e sintomas da síndrome de STH, de clínicas é derivado de inflamação não específica do ápice orbital, a fissura orbital e seio cavernoso (Buedo Rubio-Martin Tamayo Blazquez e Onsurbe Ramirez, 2015).
Por sua vez, este processo patológico tem a sua origem em um envolvimento dos nervos cranianos III, VI, IV e V (Buedo Rubio, Martin-Tamayo Onsurbe Blazquez e Ramirez, 2015).
No entanto, pesquisas atuais ainda não identificaram a etiologia específica (Buedo Rubio, Martín-Tamayo Blázquez e Onsurbe Ramírez, 2015).
Além disso, alguns relatos clínicos apontam os fatores autoimunes como a origem da síndrome de Tolosa Hunt (Martínez, Casasco, Pendre, De Bonis e Berner, 2010).
Algumas das análises sorológicas das pessoas afetadas mostraram a presença de anticorpos lúpicos, ANCA e antiperoxidase (Martínez, Casasco, Pendre, De Bonis e Berner, 2010).
Além disso, outras possíveis causas são (Organização Nacional para Desordens Raras, 2016):
- Inflamação granulomatosa.
- Inflamação generalizada dos vasos sanguíneos cranianos.
Diagnóstico
Como boa parte das patologias, no diagnóstico da síndrome de Tolosa Hunt, a análise da história familiar e individual e do exame físico é fundamental para determinar as características clínicas do paciente acometido.
Para categorizar os sinais e sintomas clínicos, os critérios propostos pela Classificação Internacional de Dores de Cefaléia Segunda Edição (Díaz, Aedo e González Hernández, 2009) são geralmente utilizados.
- Um ou mais episódios de dor localizados em áreas orbitais no nível unilateral. Apresenta um curso persistente na ausência de tratamento médico.
- Paralisia parcial ou completa de um ou mais dos seguintes nervos cranianos: par II, par IV, par VI ou identificação de granuloma através de biópsia de pele ou ressonância magnética (MRI).
- Os episódios de dor e paresia devem ser resolvidos após 72 horas da aplicação de um tratamento adequado.
- As causas etiológicas associadas a tumores, vasculites, sarcoidose, enxaquecas, diabetes ou meningite basal são excluídas.
Além disso, para o exame preciso dessas características, alguns testes de laboratório são normalmente utilizados.
Na síndrome de Tolosa Hunt, a ressonância magnética nuclear (RMN) é a técnica de lição para o estudo da inflamação do seio cavernoso (Diáz, Aedo e González Hernández, 2009).
Tratamento
Uma vez feito o diagnóstico diferencial, a abordagem terapêutica clássica é a administração de drogas corticoides.
Embora em alguns afetados seja possível que haja uma remissão espontânea dos sintomas. Se um tratamento com corticosteróides não for usado, essa condição médica pode persistir com o tempo.
O prognóstico médico é favorável. A resolução dos episódios de dor geralmente é completa com ou sem tratamento específico, embora a remissão seja comum.
Até 40% dos afetados descrevem ter sofrido vários episódios de dor orbital.
Além disso, em alguns casos, algumas alterações oculares podem persistir, especialmente aquelas relacionadas à motilidade palpebral e ocular.
Referências
- Aguirre, D., Zúñiga, G., e Barrera, L. (2014). Síndrome de Tolosa-Hunt: relato de caso e revisão de literatura. Acta Neurol Colomb.
- Buedo Rubio, M., Martín-Tamayo Blázquez, M., & Onsurbe Ramírez, I. (2015). Síndrome de Tolosa-Hunt, um diagnóstico de exclusão. Rev Pediatr. Aten Primário.
- Díaz, C., Aedo, I. e González-Hernández, J. (2009). Síndrome de Tolosa Hunt: revisão baseada em um caso clínico. Revistqa Memoriza.
- Díez de los Ríos González, A., Gómez Rebollo, C., e Aguilar Cuevas, R. (2013). Solução de casos 46. Síndrome de Tolosa-Hunt. Radiologia.
- Granados-Reyes, G., Soriano- Redondo, E. e Durán-Ferreras, E. (2012). Síndrome de Tolosa-Hunt após traumatismo ocular. Rev Neurol.
- Martínez, D., Casasco, J., Pendre, N., de Bonis, C., & Berner, S. (2010). SÍNDROME DA CAÇA DE TOLOSA. Rev Argent Neuroc.
- NORD (2016). Síndrome de Tolosa Hunt. Retirado da Organização Nacional para Desordens Raras.
- Sánchez Iñigo, L. e Navarro González, D. (2014). Síndrome de Tolosa-Hunt, mais uma dor de cabeça. Neurol Arg.
- Taylor, D. (2015). Síndrome de Tolosa-Hunt. Obtido do Medscape.
- Zimmermann-Paiz, M. (2008). Síndrome de Tolosa-Hunt precedida por paralisia facial. Relatório de um caso ... Rev Oftalmol Mex.